

Содержание
Что такое синдром Крузона
Синдром Крузона (черепно-лицевой дизостоз) — редкое генетическое заболевание, при котором кости черепа срастаются раньше времени, что приводит к деформации головы и лица.
Впервые заболевание было описано в 1912 году французским врачом Октавом Крузоном. Патология наследуется по аутосомно-доминантному типу, но около половины всех случаев вызваны случайными, спонтанными мутациями. Синдром встречается у 1 из 50–60 тысяч новорожденных и с равной вероятностью поражает как мальчиков, так и девочек.
Первые признаки могут быть заметны уже при рождении, но обычно становятся более выраженными в течение первых лет жизни ребенка. Если форма головы у малыша кажется необычной, глаза выглядят слишком выпуклыми или широко расставленными, а черты лица имеют специфические особенности, — это повод для полноценного обследования. Своевременно оказанная помощь способна предотвратить тяжелые осложнения и подарить ребенку шанс на полноценное развитие.
Причины синдрома Крузона
Синдром Крузона имеет четкую генетическую природу. В большинстве случаев (классическая форма) он вызван мутациями в гене FGFR2 на 10-й хромосоме. Данный ген отвечает за производство белка–рецептора фактора роста фибробластов 2, который играет ключевую роль в формировании и росте костной ткани. Мутация приводит к гиперактивации рецептора, что и вызывает ускоренное и преждевременное окостенение черепных швов — краниосиностоз.
Реже встречается синдром Крузона с черным акантозом (CAN), который вызывается специфической мутацией в другом гене — FGFR3. Помимо типичных черепно-лицевых аномалий, для этой формы характерны кожные проявления: гиперкератоз (утолщение кожи), бородавчатые разрастания и усиленная пигментация в складках тела.
Механизм наследования синдрома Крузона — аутосомно-доминантный. Это означает, что для передачи заболевания ребенку достаточно одной измененной копии гена от одного из родителей. Однако примерно в 50–60% случаев мутация возникает спорадически, то есть впервые у ребенка, чьи родители здоровы. Отмечается, что риск спонтанной мутации может повышаться с возрастом отца.

Симптомы синдрома Крузона
Преждевременное сращение одного или нескольких костных швов черепа в период активного роста мозга препятствует нормальному развитию головы и «запускает» цепочку последствий, приводя к характерным деформациям как мозгового, так и лицевого черепа. Клиническая картина синдрома Крузона весьма узнаваема и затрагивает не только внешность, но и жизненно важные функции.
Лицевые и черепные признаки (становятся более заметны к 3–4 годам)
- Краниосиностоз. Чаще всего преждевременно срастаются венечные швы, что приводит к формированию брахицефалии — короткой и широкой головы. Реже наблюдаются другие формы, такие как скафоцефалия (увеличение черепа в передне-заднем диаметре, когда голова становится вытянутой овальной формы) или тригоноцефалия (килевидная деформация черепа в виде треугольника).
- Гипоплазия средней зоны лица. Недоразвитие верхней челюсти и скуловых костей, из-за чего лицо кажется вогнутым в средней части.
- Глазные симптомы. Из-за маленького размера глазниц возникает экзофтальм (выпученные глаза) и гипертелоризм (широко расставленные глаза). Часто развивается расходящееся косоглазие.
- Другие особенности. «Клювовидный» нос из-за недоразвития носовых костей, низко расположенные ушные раковины и др.
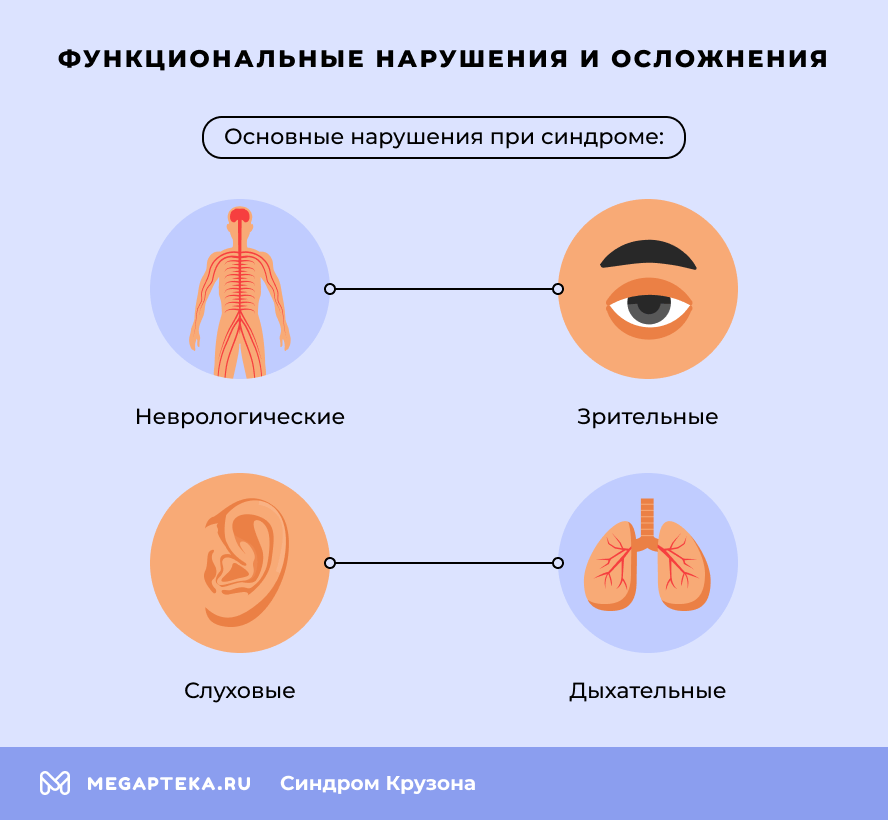
Функциональные нарушения и осложнения
- Неврологические. Повышенное внутричерепное давление из-за сдавления растущего мозга, что может привести к головным болям, рвоте, судорогам, нарушению координации движений и моторики, гидроцефалия, задержка умственного развития (без надлежащего лечения).
- Зрительные. Из-за экзофтальма возникает ксероз (сухость) и изъязвление роговицы, возможны повреждение зрительного нерва и потеря зрения.
- Слуховые. Стеноз (сужение) или атрезия (заращение) слуховых проходов, ведущие к кондуктивной тугоухости.
- Дыхательные. Вследствие гипоплазии лица и атрезии хоан (отверстий, соединяющих нос с глоткой) часто развиваются обструкция дыхательных путей, апноэ во сне.

Диагностика
Диагноз часто предполагается на основании характерного внешнего вида ребенка при рождении или в раннем младенчестве. Для подтверждения и планирования лечения необходим междисциплинарный подход с участием педиатра, генетика, нейрохирурга, челюстно-лицевого хирурга, офтальмолога и ЛОР-врача.
Ключевые методы диагностики:
- Клинический осмотр и сбор семейного анамнеза.
- Визуализация: рентгенография, компьютерная (КТ) или магнитно-резонансная (МРТ) томография черепа, которые позволяют точно оценить состояние швов, степень деформации, размеры глазниц и признаки внутричерепной гипертензии.
- Молекулярно-генетический анализ: выявление мутации в генах FGFR2 или FGFR3 является «золотым стандартом» для окончательного подтверждения диагноза.
Лечение
Специфического лекарства от синдрома Крузона не существует. Все методы лечения направлены на устранение симптомов, предотвращение осложнений и коррекцию косметических дефектов. Основным и наиболее эффективным методом является хирургическое вмешательство, которое проводится в несколько этапов по мере роста ребенка. Этапы хирургического лечения:
| Возраст ребенка | Цель хирургического этапа |
|---|---|
| От рождения до 6 месяцев | Декомпрессия и расширение черепа для снижения внутричерепного давления |
| От 6 месяцев до 3 лет | Реконструкция лобной области и глазниц для защиты глаз |
| От 4 до 12 лет | Коррекция недоразвития средней зоны лица (челюстно-лицевые операции) |
| Подростковый возраст (после 13 лет) | Ортодонтическое лечение и окончательная коррекция прикуса (ортогнатическая хирургия) |

Виды операций
- Краниопластика/фронто-орбитальное выдвижение. Расширение черепной коробки и выдвижение лобной кости и верхнего края глазниц вперед.
- Дистракционный остеогенез. Постепенное растяжение костных фрагментов для удлинения челюстей или других лицевых костей.
- Шунтирование. Установка системы для отвода избытка спинномозговой жидкости при гидроцефалии.
- Трахеостомия. Создание отверстия в трахее для облегчения дыхания при тяжелой обструкции дыхательных путей.
Консервативная и поддерживающая терапия также важна и включает наблюдение у профильных специалистов, ноотропные и сосудистые препараты (по показаниям), помощь логопеда, физиотерапевта, психолога и ношение специальных шлемов в легких случаях.

Прогноз и профилактика
Прогноз напрямую зависит от своевременности и полноты хирургического лечения. При раннем и адекватном вмешательстве, направленном на снижение внутричерепного давления и коррекцию деформаций, дети могут иметь нормальную продолжительность жизни, интеллект и удовлетворительную социальную адаптацию.
Без лечения заболевание прогрессирует, приводя к тяжелой инвалидизации: слепоте, глухоте, умственной отсталости и дыхательной недостаточности.
Профилактика как таковая не разработана из-за генетической природы болезни. Единственным методом профилактики рождения ребенка с синдромом Крузона является медико-генетическое консультирование семей, где есть случаи этого заболевания, и проведение пренатальной диагностики (биопсия ворсин хориона, амниоцентез) при планировании беременности.
Синдром Крузона — это серьезный, но управляемый диагноз. Главная опасность заключается не в самом факте генетической мутации, а в тех вторичных осложнениях, которые развиваются из-за преждевременного сращения швов черепа. Современная медицина предлагает четкий и эффективный алгоритм действий: ранняя диагностика, междисциплинарное наблюдение и поэтапное хирургическое лечение. Такой подход позволяет не просто спасти жизнь, а обеспечить ребенку с синдромом Крузона к 20 годам нормотипичную внешность и возможность расти, развиваться и жить полноценной жизнью.

Источники
Задайте вопрос эксперту по теме статьи



