

Автор статьи
Долгих Наталия Вадимовна
Профессия: провизор
Название вуза: Пермская государственная фармацевтическая академия (ПГФА)
Специальность: фармация
Стаж работы: 6,5 лет
Диплом о фармацевтическом образовании: 105924 3510859 рег. номер 31944
Места работы: провизор в аптеке, работа в офисе по снабжению аптек, провизор сервиса Мегаптека
Содержание
Ожирение — бич XXI века. Но не всегда это заболевание связано с перееданием, гиподинамией и нарушением метаболизма. Есть причины генетически обусловленного развития ожирения, например, синдром Прадера-Вилли (СПВ). Разбираем в статье, что это такое, чем отличается от синдрома Ангельмана, как болезнь проявляется, диагностируется, лечится или нет, сколько живут с СПВ.
Синдром Прадера-Вилли — довольно редкое генетическое заболевание, его частота встречаемости — один случай на 15-25 тысяч родов. Впервые синдром был описан швейцарскими педиатрами Андреа Прадером и Генрихом Вилли в 1956 году. Возникает при врожденной аномалии (отсутствии в отцовской копии) участка 15 хромосомы и встречается в равной мере у детей обоих полов. Обращаем внимание наших читателей, что мутация в генах при данном заболевании происходит случайным (спорадическим) образом и не передается по наследству.
А вот мутация в материнской копии одного гена 15 хромосомы приводит к рождению ребенка с синдромом Ангельмана. Простыми словами, синдром Прадера-Вилли возникает при дефекте хромосомы 15 отцовского происхождения, а синдром Ангельмана — хромосомы 15 по линии матери. При этом, несмотря на то, что мутация касается одной и той же хромосомной области, оба заболевания имеют различные клинические проявления.
Не уверены, какой препарат нужен? В приложении есть удобный поиск и чат с провизором!
2,5 млн установок и более 60 тысяч отзывов в App Store и Google Play

Симптомы
СПВ связан с рядом признаков, включая низкий мышечный тонус при рождении, трудности с кормлением ребенка, проблемы с обучением и поведением, а также ожирение и низкий рост.
При этом заболевании во время беременности часто регистрируется ягодичное предлежание плода, а доношенные дети имеют вес ниже нормы. После рождения и до года для детей с СПВ характерны следующие признаки:
- Гипотония: «вялый» ребенок, мышечная слабость или низкий мышечный тонус присутствуют уже при рождении.
- Трудности с кормлением: грудные дети часто испытывают трудности с грудным кормлением из-за слабости сосания, что приводит к плохому набору веса.
- Задержка развития: большинство детей с синдромом Прадера-Вилли развиваются медленнее, чем их сверстники.
- Булимия: ребенок постоянно испытывает чувство голода и много ест, в результате чего возникает ожирение, при этом жир откладывается на туловище, бедрах и плечах.
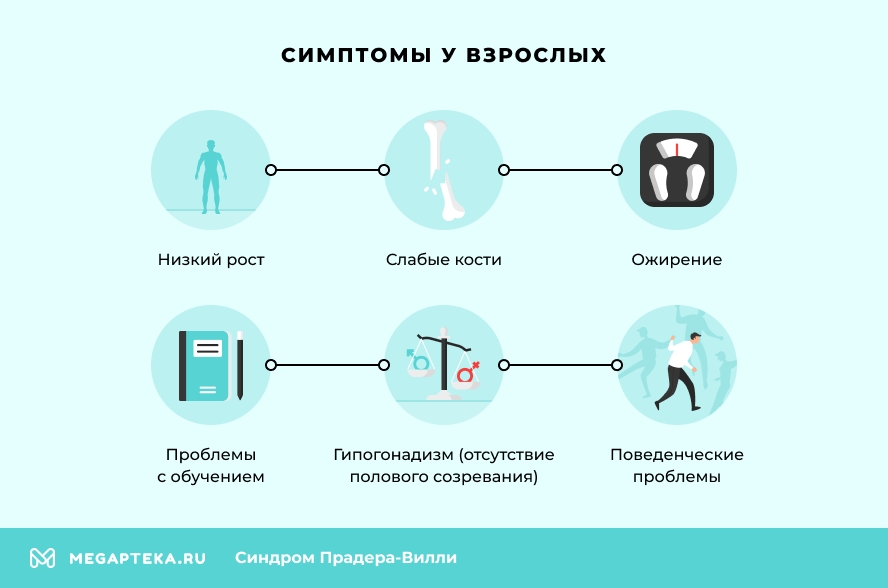
У 5-10% пациентов возникают психические заболевания (обсессивно-компульсивные нарушения, психозы), а к специфическим признакам синдрома относятся высокий болевой порог, очень вязкая слюна и высокий порог к рвоте. Другие симптомы СПВ у взрослых:
- Акромикрия: конечности (стопы и кисти) выглядят маленькими по отношению к другим частям тела.
- Низкий рост: у большинства людей рост ниже среднего.
- Поведенческие проблемы: люди с этим заболеванием проявляют агрессию, раздражительность и импульсивность.
- Ожирение: пациенты страдают ожирением уже в детском, а особенно, в подростковом возрасте и во взрослой жизни.
- Проблемы с обучением: могут испытывать проблемы с обучением, особенно в областях, требующих абстрактного мышления.
- Гипогонадизм (отсутствие полового созревания): у мальчиков гипоплазия полового члена и крипторхизм (аномально малые размеры члена и неопущение яичка), у девочек — недоразвитие больших и малых половых губ
- Слабые кости: развивается остеопороз и увеличивается риск переломов.

2,5 млн установок и более 60 тысяч отзывов в App Store и Google Play
Диагностика
Диагноз синдрома Прадера-Вилли обычно основывается на физических признаках и истории болезни пациента. Однако для подтверждения диагноза могут потребоваться дополнительные тесты, включая генетическое тестирование. Генетическое тестирование может определить, есть ли у пациента мутация в определенных генах, которые вызывают СПВ.
Обследование для исключения синдрома Прадера-Вилли проводят детям с ожирением, задержкой психического развития, мышечной гипотонией, ненормально сильным постоянным чувством голода, а также имевшим в анамнезе нарушения пищевого поведения и слабый сосательный рефлекс.

2,5 млн установок и более 60 тысяч отзывов в App Store и Google Play
Клинические рекомендации
Заболевание является генетической мутацией, поэтому вылечить синдром Прадера-Вилли нельзя. Но ранняя диагностика, терапия и специализированный уход помогают обеспечить полноценную жизнь таким пациентам.
С момента постановки диагноза (чем раньше — тем лучше) пациентам назначается препараты гормона роста, которые положительно влияют на рост, вес, мышечную силу, активность ребенка. При этом обязательно нужно соблюдать диету, поэтому может понадобиться помощь диетолога. Пациенты с синдромом Прадера — Вилли должны находиться под пристальным наблюдением эндокринолога на протяжении всей жизни.
Лечение синдрома обычно включает в себя комбинацию медицинского лечения, физиотерапии и психологической помощи. Важно начать лечение как можно раньше, чтобы максимизировать потенциал для улучшения состояния пациента. Раннее вмешательство может помочь предотвратить или уменьшить некоторые из долгосрочных последствий синдрома Прадера-Вилли.
Продолжительность жизни пациентов с синдромом Прадера-Вилли, которым вовремя диагностировали болезнь, и получившим адекватное лечение, достигает 60-70 лет. При отсутствии лечения ребенок может погибнуть даже в 4-5 лет — наиболее распространенными причинами смерти становится легочное сердце, связанное с ожирением, и дыхательная недостаточность.
Знаете, где ближайшая аптека с нужным препаратом? Приложение покажет аптеки рядом и их цены.
2,5 млн установок и более 60 тысяч отзывов в App Store и Google Play
Краткое содержание
- Симптомы могут варьироваться от человека к человеку, но общие признаки включают гипотонию, трудности с кормлением, проблемы с обучением и поведением, ожирение и низкий рост.
- Диагноз обычно основывается на физических признаках и истории болезни пациента, хотя для подтверждения диагноза может потребоваться генетическое тестирование.
- Заболевание является генетической мутацией, поэтому вылечить синдром Прадера-Вилли нельзя. Но ранняя диагностика, терапия и специализированный уход помогают обеспечить полноценную жизнь таким пациентам до 70 лет.

2,5 млн установок и более 60 тысяч отзывов в App Store и Google Play
Источники
- МедУнивер: Синдромы Прадера-Вилли и Ангельмана. Характеристика;
- Синдромы Прадера-Вилли и Ангельмана: возможности молекулярно-цитогенетической и цитогенетической диагностики. Журнал неврологии и психиатрии им. С.С. Корсакова. 2014;114(1):49‑53;
- Диагностика и лечение синдрома Прадера-Вилли. Обзор действующих международных консенсусов (дата обращения: 28.05.2024);
- Павловская Е. В., Строкова Т. В., Сурков А. Г. Синдром Прадера-Вилли // Лечебное дело. 2009. №4. URL: https://cyberleninka.ru/article/n/sindrom-pradera-villi (дата обращения: 28.05.2024).




Выпускающий редактор
Асанова Наталья Геннадьевна
Эксперт-провизор