
Подписывайтесь на нас в социальных сетях
и читайте полезные статьи о здоровье каждую неделю

Автор статьи
Гришина Александра Николаевна,
Врач-терапевт
Содержание статьи
Синдром Марфана – наследственная аномалия, для которой характерно системное поражение соединительной ткани. Заболевание проявляется изменениями в строении скелета, офтальмологическими патологиями, нарушениями функции сердца и дыхания. Предотвратить или вылечить болезнь невозможно. Но правильно подобранное лечение помогает спасти пациентов от многих осложнений и продлить им жизнь.
Причины синдрома
В развитии болезни «виновата» генетика. Синдром вызывается мутациями в гене FBN1, отвечающего за выработку фибриллина. Этот белок несет ответственность за прочность и эластичность соединительной ткани. Потеря соединительнотканными структурами своих главных свойств и становится причиной всех негативных изменений в организме.
Наследуется дефект от родителей, страдающих проявлениями синдрома. Шанс получить его у их детей составляет примерно 50%. Болезнь не передается через поколение: если у больных родителей родился здоровый ребенок, своим потомкам дефектный ген он не передаст.
Однако в 25% случаев синдром развивается у ребенка, рожденного от здоровых родителей. И конкретных факторов, вызывающих развитие синдрома Марфана, до сих пор не установлено.
Признаки мутации могут проявиться уже в первые месяцы жизни. Но нередко проявления болезни стерты и диагноз устанавливается уже во взрослом возрасте.
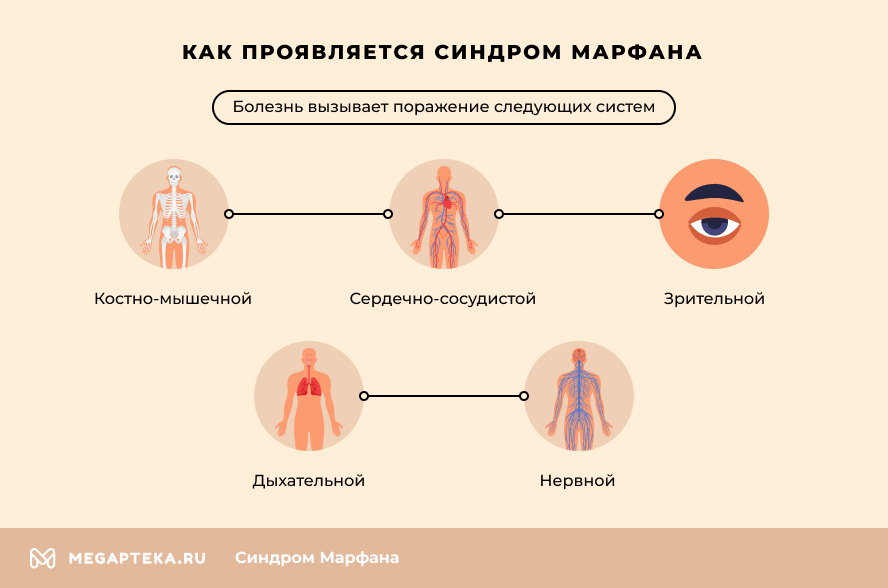
Как проявляется синдром
При стертой форме болезни поражаются 1-2 системы организма (например, скелетно-мышечная и/или органы зрения), а симптоматика проявляется незначительно. Несмотря на нарушения, такие пациенты живут практически нормальной жизнью. При выраженной форме поражаются 3 и более систем организма, либо наблюдаются значительные изменения в одной из систем.
Один и тот же генетический дефект проявляется по-разному – от незначительных изменений до тяжелых нарушений работы органов.
Болезнь вызывает поражение:
- Костно-мышечной системы – для пациентов характерен высокий рост, патологическая худоба, удлиненное лицо. Другие яркие симптомы – удлиненные и тонкие пальцы, нестандартная длина рук. Осанка в большинстве случаев нарушена, определяются сколиоз или кифоз. Нередко у пациентов наблюдается плоскостопие и повышенная подвижность суставов.
- Сердечно-сосудистой системы (ССС) – синдром вызывает нарушения сердечного ритма, недостаточность митрального клапана, различные пороки сердца. Особая опасность кроется в патологических изменениях в аорте. У большинства пациентов повышен риск расширения ее восходящей части и клапанного кольца, формирования аневризмы.
- Зрения – чаще всего наблюдается выраженная близорукость, подвывих хрусталика или изменение его положения. Также существует повышенный риск отслойки сетчатки. Нередко у больных уже в молодом возрасте развиваются глаукома и катаракта.
- Дыхательной системы – патологическое разрастание соединительной ткани в легких вызывает сужение бронхов и легочный фиброз. Нередко генетическая мутация приводит к развитию бронхиальной астмы.
- Нервной системы – в большинстве случаев нарушения мозговых структур отсутствуют. Но расширение соединительнотканной капсулы вокруг спинного мозга провоцирует нарушения движений ног, функции мочевого пузыря и кишечника. Высокий выброс адреналина часто вызывает повышенное нервное возбуждение и гиперактивность.
Вам может быть интересно: Как проявляется Синдром Гийена-Барре?
Также при синдроме Марфана могут наблюдаться опущение почек, матки, варикоз, патологии желудочно-кишечного тракта (ЖКТ).
Но физические недостатки природа компенсирует высоким уровнем умственных способностей. Нередко у таких пациентов выявляются неординарное мышление и повышенный интеллект.
Симптомы
Диагностика
Диагноз устанавливается на основании:
- семейного анамнеза – уточнение данных о состоянии здоровья родителей;
- физикальной диагностики – наличие типичных признаков, в том числе, особенности антропометрических показателей;
- данных исследований – ЭКГ (электрокардиография), ЭхоКГ (эхокардиография) сердца, МРТ (магнитно-резонансная томография) мозга и позвоночника, рентгенологического, офтальмологического обследований;
- генетического обследования.
Лечение
Лекарственных методов терапии этой патологии, к сожалению, пока не разработано. Лечебные мероприятия зависят от конкретных проявлений болезни.
Если нарушения ССС выражены не сильно, назначают консервативную терапию. При значительном расширении восходящей части и расслоении аорты, пролапсе митрального клапана, пороках сердца применяют хирургические методы.
Коррекция зрения проводится с помощью очков и контактных линз. При необходимости выполняется хирургическое лечение глаукомы, катаракты, замена смещенного хрусталика искусственным.
Для поддержания опорно-двигательного аппарата применяется коллаген-нормализующая терапия, назначаются витаминно-минеральные комплексы. Выраженные скелетные нарушения корректируются хирургическим путем.
Клинические рекомендации исключают для пациентов с мутацией физические нагрузки, травмоопасные игры, высокую активность. Для поддержки позвоночника показано ношение корсета, с целью укрепления мышц – занятия ЛФК (лечебная физическая культура) и массаж.
Жизнь людей с этой генетической мутацией нельзя назвать простой. Но при ответственном отношении пациентов к своему здоровью уменьшить проявление болезни и предупредить серьезные осложнения вполне реально.

Поделиться мегасоветом
Понравилась статья? Расскажите маме, папе, бабушке и тете Гале из третьего подъезда